
A decade ago, I began my PhD at
Vanderbilt University in Nashville, Tennessee, where I was interested in
studying the evolutionary process of speciation (or how new biological species
evolve). I was very lucky during my PhD to be surrounded by great people.
Case in point, I shared an office for part of the year with a visiting
collaborator, Patrik Nosil, who studied speciation in a group of stick insects
called Timema. Second, my PhD advisor
encouraged me to invite great thinkers on speciation to be part of my
dissertation committee – enter Jeff Feder from the University of Notre Dame,
who studied speciation in a group of fruit-feeding flies called Rhagoletis and served as my external
committee member. These connections made during the beginning of my PhD last to
this day.
 |
| Figure 1. The Pancake Pantry in Nashville, TN, USA. |
During a fateful visit to a common grad
student hangout (circa 2007), the Pancake Pantry (Fig. 1), Patrik Nosil and I and a group of graduate students
started discussing the age-old debate about the number of genes involved in
adaptation (and speciation): few versus many? And whether the traits
responsible for adaptation and speciation were polygenic traits or traits with
a simple genetic basis? One way we thought to test this was to use as many
molecular markers as you could survey, distributed across the genome, and ask
the question: how many of these gene regions exhibit significant population differentiation,
but are restricted to populations adapting to different environments? We came
up with ideas of how to test it, and what type of tools we would need, right
over our plates of pancakes! I think we even had a budget by the time we walked
back in our calorie coma from lunch. My
major takeaway from this lunch was that I now considered the genome as an
active player, not a passive mediator, in the speciation process and I would
never think about speciation in the same way again!
What emerged initially from this pursuit were two comparative AFLP genome scans of two different study systems, each
undergoing speciation driven by divergent ecology, that were published in the
journal Evolution (Nosil et al. 2008; Egan et al. 2008). These studies were very informative in
highlighting the proportion of gene regions (AFLPs) in the genome exhibiting strong
differentiation between divergent populations, and possibly addressed the
repeatability of gene regions associated with adaptation to two environments
(in our case, host plants). But we were
also left with many more questions than answers. How were these divergent loci
distributed and arrayed across the genome? And were the loci exhibiting strong differentiation
driven by selection or other evolutionary phenomena?
Fast-forward to 2010 – I finished my PhD and
I was awarded a Faculty Fellowship at the University of Notre Dame, which came
with some seed money for research and the chance to work more closely with my
external committee member, Jeff Feder. Almost immediately upon arriving in
South Bend, IN, Patrik (now in Sheffield, UK), Jeff, and I had a set of
conference calls and email exchanges that started the project that would result
in the Ecology Letters MS I will summarize below. (Jeff and Patrik had just
finished a sabbatical in Berlin the year before where they spent much of their
time ruminating on the genome-level phenomena influencing the speciation
process.) We recruited other evolutionary biologists well trained in Rhagoletis biology (Tom Powell, Glen
Hood, and Greg Ragland), as well as two computer scientists (Scott Emrich and
his PhD student Lauren Assour) with the ability to process the large amount of
data we would gather.
Our interests were to better understand
the role the genome might play in the evolution of new species. We were
inspired by a paper published over 30 years ago by Joe Felsenstein (1981),
where he described the difficulty of building up many-locus differences between
populations if gene flow was ongoing and recombination was breaking up
associations. This conflict between selection and gene flow would form the
basis for our project. How is it that populations can diverge in the face of
ongoing gene flow? What are the properties or characteristics of species that
are suspected of speciation-with-gene-flow which facilitated their divergence?
 |
| Figure 2. Rhagoletis pomonella exploring the fruit of the hawthorn tree (Crataegus mollis). Photo credit: Hannes Schuler |
Rhagoletis
pomonella offered a
great study system to test these ideas, as it is a well-documented case of
speciation-with-gene-flow (Fig. 2). Rhagoletis
pomonella is a member of a sibling species complex containing numerous geographically
overlapping taxa proposed to have radiated in sympatry by adapting to many new
host plants from several different plant families. Rhagoletis flies infest the fruits of their host plants, where host
fruits are typically available for a discrete window of time over the growing
season and each fly species completes one generation per year. Adult flies meet
exclusively on or near the host fruits to mate; females oviposit into the host fruit;
larvae consume the fruit, then burrow into the soil to pupate, entering a
pupal diapause that lasts until the following year. Thus, phenological matching
of fly to host-plant fruiting is critical to fly fitness.
The most recent example of a host shift
driving speciation is the shift of R.
pomonella from its native host hawthorn to introduced, domesticated apple,
which occurred in the mid-1800’s in the eastern United States. Genetic and
field studies have shown that apple and hawthorn flies represent partially
reproductively isolated host races and that gene flow has been continuous
between the fly races since their origin. One key trait that differs between
the races is the timing of diapause termination, which varies between the races
to match the 3–4 week earlier fruiting time of apple versus hawthorn trees (Fig.
3). Rhagoletis emerge from their
fruits as late-instar larvae and overwinter in the soil in a facultative pupal
diapause. The earlier fruiting time of apples therefore results in apple flies
having to withstand warmer temperatures for longer periods prior to winter. As
a result, natural selection favors increased diapause intensity, or greater
recalcitrance to cues that trigger premature diapause termination in apple
flies.
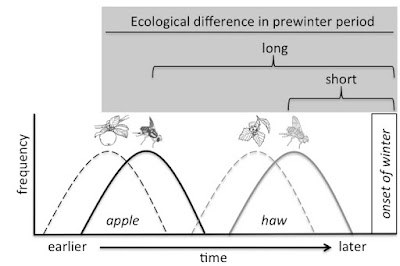 |
| Figure 3. Fruit on apple trees ripens 3-4 weeks earlier than hawthorn fruit (dashed lines). Apple flies eclose earlier as adults (solid lines) and are exposed to warmer temperatures as pupae in the soil for a longer period of time before winter. |
Jeff had the perfect experiment frozen in
his freezer from 20 years ago. Previously, his lab had reared the ancestral haw
race of Rhagoletis under the phenological
conditions of both host plants it attacks in nature. He had previously looked
at changes in a set of allozymes and microsatellites, but did not have the ability at the time to look across
the genome at tens of thousands of SNPs. Specifically, he exposed ancestral hawthorn
fly pupae to warm temperatures for a short 7-day (‘hawthorn-like’ control) vs.
long 32-day (‘apple-like’ experimental) period prior to winter (Fig. 4).
 |
| Figure 4. In the selection experiment, hawthorn flies were exposed to a short (7-day) versus long (32-day) prewinter period to emulate the time difference experienced by hawthorn versus apple-fly pupae in nature. |
We also had a
specific hypothesis we wanted to test that integrated Jeff’s selection
experiment with sampling from natural populations. We tested whether the
changes across the genome induced by the lab experiment on divergent host-plant
phenology would predict the genome-wide differences observed at these same loci
between natural sympatric populations. In this experiment, we stressed that we
were quantifying the total genome-wide impact of selection, which involves both
direct effects, where natural selection favors the causal variants underlying
selected traits, and indirect effects, where additional loci respond because
they are correlated due to linkage disequilibrium with these causal variants. Thus,
the ‘total’ impact of divergent selection (i.e. direct + indirect effects) that
we quantify here can involve changes at many loci (Gompert et al. 2014;
Soria-Carrasco et al. 2014).
Quantifying the
impact of selection genome-wide is important because, as populations diverge,
the effects that individual genes have on reproductive isolation (RI) can
become coupled, strengthening barriers to gene flow and promoting speciation (Barton
1983, Bierne et al. 2011). If predicated solely on new mutations, this
transition could take a long time and populations could go extinct or
conditions change without speciation, which may explain why sympatric speciation
is difficult to observe and test. Thus, a prediction for systems with the
potential for speciation-with-gene-flow is that they exhibit large stores of
standing variation and consequently, show extensive, genome-wide responses to
selection when challenged by divergent ecology.
In our selection
experiment, about 6% of the SNPs showed significant frequency shifts between
the short and long prewinter periods. However, because of extensive linkage
disequilibrium (LD) in Rhagoletis,
these SNPs did not provide an estimate of the independent number of gene
regions influenced by selection. Thus, we assessed the pattern of LD between
SNPs to delimit independent sets of loci.
We determined that the 6% of responding SNPs represented 162 different
sets whose members were in LD with each other, but in equilibrium with all
other SNPs. After accounting for the table-wide null expectation of 52
significant sets due to type I error, using a modeling approach we detail in our
Supplemental material, a lower bound estimate of 110 gene regions responded to
selection. To determine how physically widespread the response was across the
genome, we constructed a recombination linkage map for Rhagoletis that contained 2,352 SNPs. About 13% of mapped SNPs
showed significant frequency shifts in the selection experiment and were
dispersed widely across the five major chromosomes of the R. pomonella genome (Fig. 5). Thus, numerous independent gene
regions responded to selection and they were distributed throughout the genome.
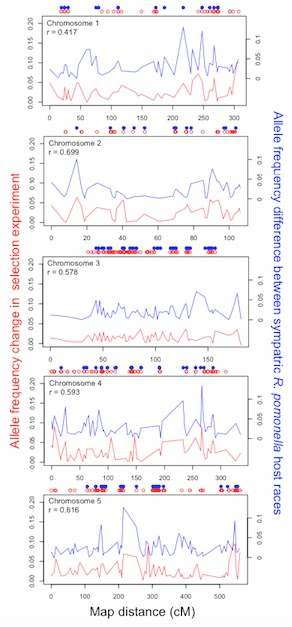 |
| Figure 5. Genome-wide comparison of allele frequency shifts in the selection experiment (red line; left axis) versus divergence between field-collected sympatric host races (blue line; right axis) along chromosomes 1-5. Circles above panels denote SNPs showing statistically significant response in the selection experiment (open red) or difference between the host races (solid blue). Correlation coefficient (r) is reported independently for each chromosome. |
Now we tested
our main hypothesis: does the genomic response in the selection experiment
reflect nature? The answer is yes. The
direction and magnitude of allele frequency changes for all 32,455 SNPs in the
selection experiment was highly predictive of genetic differences between the
sympatric hawthorn and apple host races at the Grant, MI, site (r = 0.39, P <
10-6). Most strikingly, for the SNPs showing significant responses in
both our selection experiment and host divergence in nature, the allele that
increased in frequency in the hawthorn race after selection was the exact same
allele in higher frequency in the apple race in nature (P = (½)154 =
4.4x10-47).
To what extent
did the single bout of selection on hawthorn flies genetically create the
derived apple race? The answer is a good
deal. For all 32,455 SNPs, the mean SNP frequency for hawthorn flies surviving
the long prewinter treatment shifted 38.9% of the difference between the host races
toward apple flies. For the 154 SNPs showing significant responses in
the selection experiment and host divergence, the shift was 84.1%.
Why is the
impact of divergent ecological adaptation so pronounced and pervasive in Rhagoletis? One contributing factor is the extensive LD
in the fly, some of which is due to inversions, requiring additional DNA
sequence analysis to resolve. A second factor is the presence of substantial
standing genetic variation in R.
pomonella, which supports the hypothesis that such stores may define taxa
having a greater capacity for speciation-with-gene-flow. Finally, when
ecological adaptation involves traits like diapause that can be highly
polygenic, selection may more often have genome-wide consequences. In this
regard, microarray studies of R.
pomonella have revealed hundreds of loci varying in expression during
diapause breakage that are potential targets of selection (Ragland
et al. 2011).
 |
| Figure 6. Rhagoletis pomonella fly exploring apple fruit. Photo credit: Andrew Forbes |
Interestingly,
this work shares some important similarities and differences with other recent
studies combining selection experiments with surveys of genome-wide genetic
variation in natural populations, including the Timema ecotypes that are the mainstay of the Nosil lab. In both a
within-generation (Gompert et al. 2014; similar to the Rhagoletis study here) and a between-generation study of selection
in the field (Soria-Carrasco et al. 2014), a genome-wide response involving
many loci was observed. However, LD was much lower in the Timema ecotypes, and thus the association between genetic differences
induced in those selection experiments did not match natural genetic variation
as closely as in the Rhagoletis
experiment.
In summary,
divergent ecological selection can have genome-wide effects even at early
stages of speciation. Large stores of standing variation in Rhagoletis flies may
potentiate the evolution of genome-wide reproductive isolation and their
adaptive radiation with gene flow. As the study of speciation genomics expands,
it will be possible to test the degree to which other taxa prone to ecological
sympatric speciation share similar characteristics as R. pomonella, and to assess the relationship
between standing variation and clade richness.
That was one
productive plate of pancakes!
References:
Barton,
N.H. 1983. Multilocus clines. Evolution
37, 454–471.
Bierne, N.,
Welch, J., Loire, E., Bonhomme, F. & David, P. 2011. The coupling hypothesis:
why genome scans may fail to map local adaptation genes. Molecular Ecology 20, 2044–2072.
Egan, S.P., P. Nosil,
& D.J. Funk. 2008. Selection and genomic differentiation during ecological
speciation: isolating the contributions of host-association via a comparative
genome scan of Neochlamisus bebbianae leaf beetles. Evolution 62: 1162-1181.
Egan, S.P., G.R.
Ragland, L. Assour, T.H.Q. Powell, G.R. Hood, S. Emrich, P. Nosil & J.L.
Feder. 2015. Experimental evidence of genome-wide impact of ecological selection
during early stages of speciation-with-gene-flow. Ecology Letters, online
early. (doi: 10.1111/ele.12460)
Felsenstein J. 1981. Skepticism towards Santa Rosalia, or why are there
so few kinds of animals? Evolution 35:124 – 138.
Gompert, Z., A.A.
Comeault, T.E. Farkas, J.L. Feder, T.L. Parchman, C.A. Buerkle, and P. Nosil. 2014. Experimental evidence
for ecological selection on genome variation in the wild. Ecology Letters 17:
369-379.
Nosil, P., S.P. Egan, & D.J. Funk. 2008.
Divergent selection plays multiple roles in generating heterogeneous genomic
differentiation between walking-stick ecotypes. Evolution 62: 316-336.
Ragland, G.J., S.P. Egan, J.L. Feder, S.H. Berlocher,
& D.A. Hahn. 2011. Developmental
trajectories of gene
expression reveal regulatory candidates for diapause termination, a key
life history transition in the apple maggot fly, Rhagoletis pomonella. Journal
of Experimental Biology 214:
3948-3960.
Soria-Carrasco,
V., Z. Gompert, A.A. Comeault, T.E. Farkas, T.L. Parchman, J.S. Johnson, C.A.
Buerkle, J.L. Feder, J. Bast, T. Schwander, S.P. Egan, B.J. Crespi, & P. Nosil. 2014. Stick insect
genomes reveal natural selection's role in parallel speciation. Science 344: 738-742.




4.4x10-47 is an impressive P-value! I wonder whether it might be the smallest P-value ever published for experimental work in ecology and evolution? Probably not, I suppose – but then what is? :-> Great post, really interesting!
ReplyDelete